
British Society of Urogynaecology

- About
- Patient Information
- Healthcare Professionals
- Donate
In 2014 there were some concerns raised in Scotland about the safety of synthetic meshes for prolapse and incontinence surgery. The MHRA have a reporting system for medical devices similar to the “yellow card” drug adverse event reporting system. To date the number of reports to the MHRA from medical practitioners of adverse events is less than would be expected. The following is a guide to reporting device complications to the MHRA.
The MHRA define an adverse incident as "an event that causes, or has the potential to cause, unexpected or unwanted effects involving the safety of device users (including patients) or other persons." In other words any actual or potential adverse event which occurs when employing a device which affects the patient, healthcare professional or other user
Any adverse incident involving a device should be reported to the MHRA, including problems with the instructions for use or difficulties with deployment, especially if the incident has led, or might have led to:
For synthetic meshes for prolapse and incontinence this may include:
These may present to the original operating surgeon but may present later to other gynaecologists, urologists or colorectal surgeons.
Adverse incidents can be reported by
NHS trusts and primary care trusts in England have all designated a member of staff as their medical device liaison officer (MDLO). Their role is to promote and coordinate adverse incident reporting and help to manage the dissemination of medical device alerts.
When a device related adverse event is reported to MHRA, the MDLO and/or risk manager should also be informed in line with local protocol.
The MHRA does not require patient names or other identifying information in order to carry out an investigation. Healthcare staff reporting incidents should, therefore, ensure that such details are deleted or redacted from their reports, from accompanying attachments and from any subsequent correspondence.
The details required by the MHRA are specified on the report forms. The reporter's full contact details (name, post held etc.) are essential, as this allows the MHRA to contact the reporter to acknowledge receipt of the report and request any further information that may be needed.
The preferred and simplest way to report adverse incidents involving devices to the MHRA is via the on-line forms on the MHRA website (http://www.mhra.gov.uk). This also provides facilities for copying the incident through to MDLOs, risk managers or other local relevant staff in compliance with any good practice in local reporting policies. You may however also fax, email or post a downloadable form instead.
Each devolved administration has its own guidance on reporting adverse incidents, available on the respective websites.
Northern Ireland (external link)
Scotland (external link)
Wales (external link
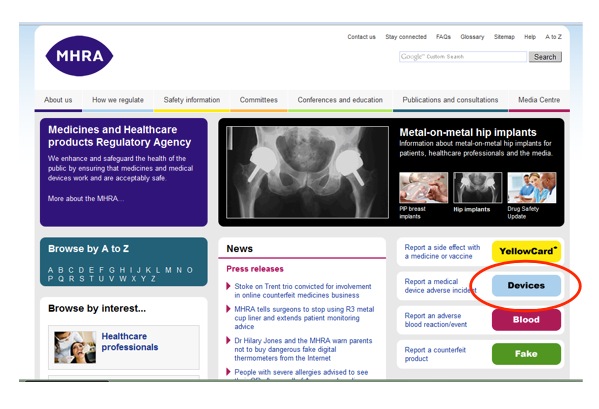

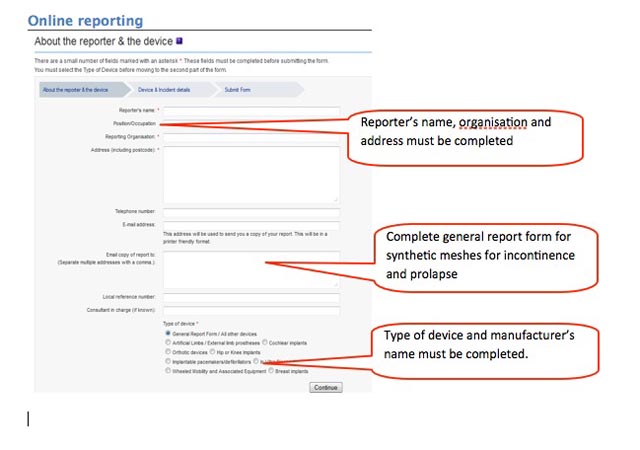
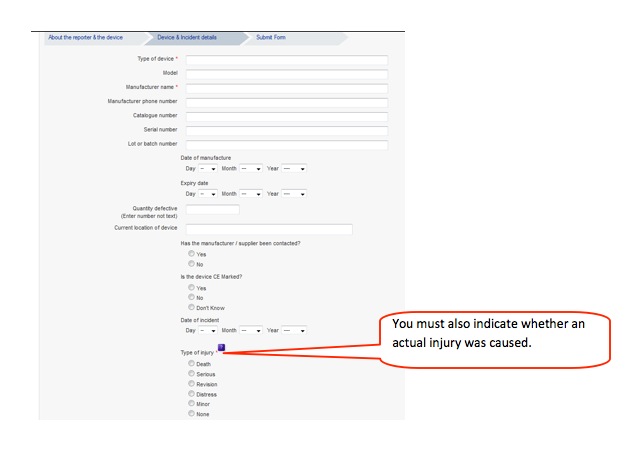
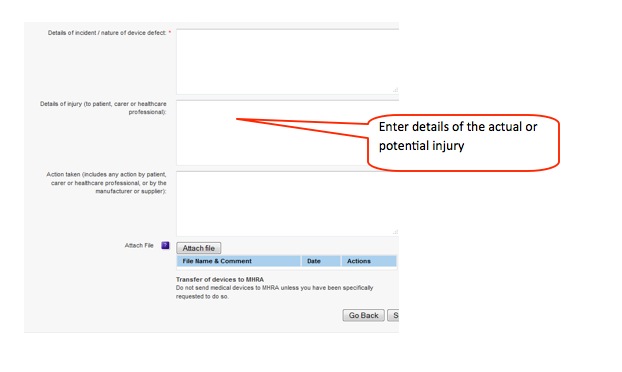
How to report an adverse incident report (England and Wales):
1. Go to MHRA website
2. Follow links to "Devices" then "Medical device adverse incident reporting forms"
3. Complete form, preferably online. The core information required is very simple:
4. If you include your own email address on the online form, MHRA will email you a pdf copy of your completed form.
5. Inform Trust medical devices liaison officer or equivalent in your Trust. You can do this electronically by also entering their email address on the online form.
Working in association with
![]()
![]()
Find us at:
BSUG
c/o Royal College of Obstetricians & Gynaecologists
10-18 Union Street
LONDON
SE1 1SZ
Registered charity No 1143157
Privacy and cookiesQuick Links
Our Members
